Discovery to Delivery
› Solutions ›Cell and Gene Therapy
Preclinical in vitro and in vivo proof-of-concept, pharmacology, and toxicology studies are conducted to establish feasibility and rationale for clinical use of the investigational cellular and gene therapy cellular and gene therapy (CGT) product, as well as to characterize the product’s safety profile. These studies also serve as the scientific basis to support the conclusion that it is reasonably safe to conduct the proposed clinical investigations. Due to the diversity of CGT products, it is important to conduct a careful benefit-risk analysis, performed in the context of the particular clinical condition under study. Preclinical data derived from studies conducted in appropriate animal species and animal models of disease contribute to defining reasonable risk for the investigational CGT product.
Preclinical studies are intended to define the pharmacologic and toxicologic effects predictive of the human response, not only prior to initiation of clinical trials, but also throughout drug development. The goals of these studies include the following: to define safe starting doses and escalation schemes for clinical trials, to identify target organs for toxicity and parameters to monitor in patients receiving these therapies, and to determine populations which may be at greater risk for toxicities of a given cellular or gene therapeutic.
Design of preclinical studies should take into consideration: 1) the population of cells to be administered or the class of vector used, 2) the animal species and physiologic state most relevant for the clinical indication and product class, and 3) the intended doses, route of administration, and treatment regimens.
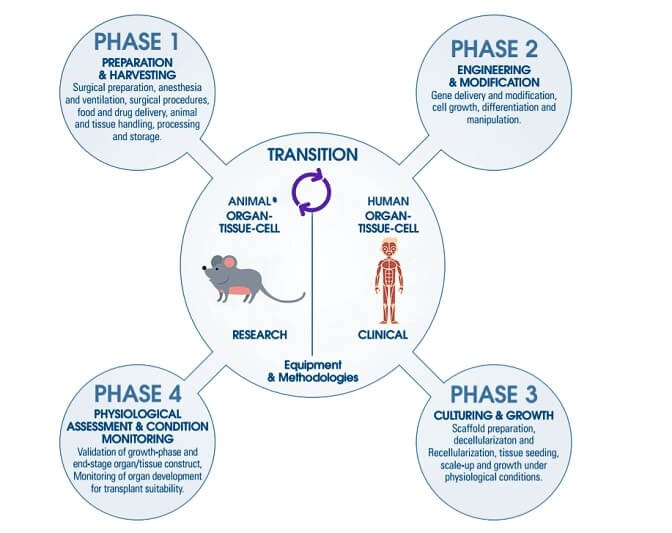
The Preparation and Harvesting phase of cell and gene therapy includes the acclimatization of the test animals for extraction. Animal workstations are necessary for containment purposes during animal handling mainly for personnel and environment protection.
With the introduction of the VIVA® range, Esco applies decades of experience in clean air technologies to the animal research laboratory, protecting personnel and the laboratory environment from:
This phase also covers up the cell collection process, hence, it is extremely important to consider the following criteria before and/or after the cell/tissue of the species have been extracted:
The transfer of genetic material can be accomplished in vivo through local or systemic inoculation or ex vivo where the target of interest is collected and modified outside of the organism before being returned to the host. Transfer of synthetic DNA can be accomplished by transduction or transfection. Such methods of transfer include either direct injection of DNA into the recipient cells, or utilization of methods to induce membranes permeabilisation, receptor-mediated uptake or endocytosis. Transduction utilizes recombinant virus as a vector for gene transfer.
While natural transfer of genetic material can be accomplished in several ways, degradation of plasmid DNA is susceptible because of biologic enzymes, hence the following synthetic delivery systems are used:
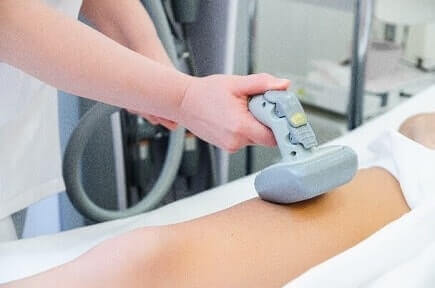
Iontophoresis is a non-invasive method of propelling high concentrations of a charged substance, normally medication or bioactive agents, transdermally by repulsive electromotive force using a small electrical charge applied to an iontophoretic chamber containing a similarly charged active agent and its vehicle.

This is a significant increase in the electrical conductivity and permeability of the cell plasma membrane caused by an extremely applied electrical field. It is usually used as a way of introducing some substance into a cell, such as loading with a molecular probe, a drug that can change the cell’s function, or a piece of coding DNA.
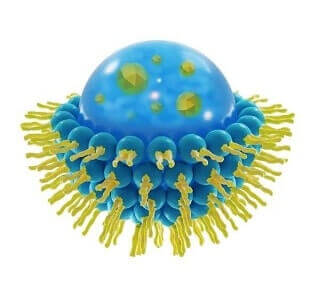
This is a spherical vesicle with a membrane composed of phospholipid and cholesterol bilayer. Liposomes can be composed of naturally-derived phospholipids with mixed lipid chains or of pure surfactants. To deliver the molecules to sites of action, the lipid bilayer can fuse with other bilayers such as the cell membrane, in order to deliver the liposome contents.
An injector is a pump-like device, which charges or discharges containers under pressure with suitable arrangements. Motive force is gained at the inlet from a suitable gas or liquid that is under pressure. Pneumatic injectors can deliver 10-15 L femtolitre of materials and specialized mechanical pumps are capable of 10-12 L picolitre delivery volumes.
Scaffolds used for tissue engineering and regeneration can be synthetic polymers, naturally derived proteins/polymers, and tissue-derived scaffolds produced by decellularization. Synthetic scaffolds can be tailored and reproduced readily with controlled mechanical properties, porosity, and surface topography. However, this approach faces limitations such as uncertainties regarding the degradation rate and products as well as cell compatibility. Naturally derived proteins/polymers (e.g. collagen, fibrin, alginate) possess intrinsic cell compatibility, but they are often weak in mechanical properties and difficult to manipulate.
Decellularization, on the other hand, removes cellular components from tissues/organs to generate extracellular matrix templates, a complex mixture of structural and functional proteins that can be used for tissue engineering applications. The removal of cellular content and antigens from the tissue-derived scaffolds reduces foreign body reaction, inflammation, and potential immune rejection. Effective decellularization methods include chemical, enzymatic, physical or combinational approaches. The working principle of these methods is through disruption of cell membrane, which will then cause the cellular contents to be released and rinsed away.
Decellularization includes chemical methods such as the utilization of ionic detergents like sodium dodecyl sulfate (SDS), non-ionic detergents (Triton X-100), chelating agent (EDTA), zwitterionic-based detergent, acids and bases, hypotonic and hypertonic solutions, alcohols and acetone and enzymatic method such as trypsin treatment. Physical method like scraping, solution agitation, pressure gradient, and freezing.

Scaffolds are sterilized afterwards, preventing contamination and potential transmission of pathogens. Sterilization involves the use of sterility test isolators and acellular scaffolds including ethanol, peracetic acid, ethylene oxide, ultraviolet and gamma radiation. Aside from sterilization, scaffold stabilization is also used to delay degradation, neutralize antigens, and stabilize the overall structure of ECM.

General Platform Processing Isolator (GPPI) is especially designed as a sterility test isolator.
Cryopreservation or lyopilization have also been both used in decellularization approaches for storage of scaffolds for easier handling. Cryopreservation is able to effectively preserve the acellular tissue scaffolds at sub-zero temperatures maintained by liquid nitrogen.

Different freeze drying applications such as preservation of biological samples, purification of proteins, and concentration of chemical products, are now done perfectly with SubliMate®.
The following criteria should also be considered during the phase III of regenerative therapy:

CelCradle™ is a disposable bioreactor capable of high-density cell culture for protein expression, virus, and monoclonal antibody production. It is designed based on the concept of bellow-induced intermittent flow of media and air through porous matrices, where cells reside. This provides a low shear, high aeration, and foam-free culture environment.
Cell culturing practices and facilities should be designed to avoid contamination of one cell culture with another.
During cell culturing, extensive drift in the properties of a cell population, or overgrowth by a different cell type originally present in low numbers, may occur. To detect such changes, cell identity should be assessed quantitatively, for example, by monitoring cell surface antigens or biochemical markers. The method of identification chosen should also be able to detect contamination or replacement by other cells in use in the facility. Acceptable limits for culture composition should be defined. Quantitative assays of functional potency may sometimes provide a method for population phenotyping. The desired function should be monitored when the cells are subjected to manipulation, and the tests carried out periodically to assure that the desired trait is retained. Identity testing should, in some cases, include verification of donor-recipient matching and immunological phenotyping.
The major progress in microsystem technologies for creating small, integrated and reliable microtransducers devices in combination with biological sensing elements has revolutionized the field of biosensors during the last decade. Such micro-biosensor systems raised the expectation to get a comprehensive insight into dynamic cellular metabolic events and subsequently a complete understanding of the metabolism of human biology.
Two different critical factors govern the aseptic processing of aseptic drugs involving cell/gene therapy. The first of these is “risk evaluation/analysis”, the goal of which is risk quantification. The other is “risk mitigation” that considers the possibility of contamination during facility design, equipment selection, definition of process, and the operation of the process itself. Risk mitigation can both indicate the level of need for process improvement and determine the priority that should be assigned to mitigation activities. Risk mitigation as it relates to aseptic processing generally means to properly control human intervention, but ultimately if risk mitigation is to be maximized, personnel must be completely removed from critical area of aseptic processing, or sterility test isolators should be utilized.
It has continually been assumed by some scientists and engineers that aseptic conditions can adequately be maintained if the personnel conducting aseptic processing wear aseptic gowns properly and exhibit sound aseptic technique in conducting their work. However, it is clear from published research data that personnel emit very high levels of microbiological contamination into the environment. This is true even when state-of-the-art gowns are chosen and worn properly. According to Ljungqvist et al., personnel wearing previously unused sterilized gowns emit 1500–3000 cfu/h of microbiological contamination even during processes involving limited operations.
The microbiological contamination released by personnel can increase to ≥10,000 cfu/h during complex operations that require the execution of many physical tasks.
The total number of aerobic bacteria existing on human skin is normally >1.2 million per square meter. The number of microorganisms existing on a healthy human’s hands and arms are in the range 0.9–3 million per square meter. Microorganisms are emitted by gowned personnel by a pumping effect of the technician’s gown during operation. Hence, advanced aseptic processing technologies such as Blow-Fill Seal and Restricted Access Barrier System on top of isolators (Compounding Aseptic Containment Isolator and Compounding Aseptic Isolator) are used to reduce the possibility of product contamination caused by personnel.

The closed RABS is an intermediate solution between isolators and open RABS. A closed operation RABS provides a higher level of contamination control because the RABS’ barrier doors remain closed from the point of the last bio-decontamination, through the initial set-up and processing. These systems typically use transfer systems which are similar to isolator type transfer systems that are closed and dock with the RABS.
Isolator systems do not allow direct interventions by personnel into the enclosure’s aseptic processing area. To ensure that contamination is not introduced into the isolator, it is critical to have means of bringing the materials into and out of the isolator in a way that doesn’t compromise and allow leakage of contaminants within the enclosure. The sterility assurance provided by an isolator is a product of physical separation of personnel, elimination of risk of introduction of airborne contamination, and systems that prevent the introduction of non-sterile materials into the isolator. Safe, contamination free transfer of materials into and out of the isolator can be assured by the use of devices known as decontamination pass boxes and circular-shaped rapid transfer ports (RTP); decontamination interfaces have also been developed for isolators.
Large volumes of air are required to properly satisfy the international standards for cleanroom air. Air handling systems such as isolators and the build and design of the cleanroom is critical as these will edict the permitted processes during the sterile compounding.
ISO Class 5/Grade A environments are required for the critical area of aseptic processing. The critical area in aseptic processing is defined as the part of a facility where product and sterilized components are brought together and filled and/or assembled. It is necessary to install and operate this ISO Class 5/Grade A critical zone environment within an ISO Class 7/Grade B environment. Equally important to this is the provision of proper gowning and support facilities to ensure safe, contaminant-free personnel and component entry. Alternatively, in some modern facilities, the entire aseptic processing room can be designed to ISO Class 5/Grade A requirements; however, such a facility is both expensive to build and operate.
The facility and the utility requirements are the fundamental support of the process flow and production effectiveness. In facility design, it is important to consider the regulatory nature of the industry and to seek some in-depth knowledge of the requirements, hence, serving the most basic foundation for a safe, effective, and pure drugs while maintaining its cost-effectiveness.
Cleanrooms are extremely critical on the manufacturing of biologics, hence the use of Cross Contamination Facility Integrated Barrier (CCFIB) can be used to maximize the performance given by a cleanroom to ensure the integrity and quality of any drug product. This equipment will minimize the exposure of chemical or biological contaminants, such as micro-organisms or adventitious agents, thus providing utmost protection to the product, operator and the environment.

The CradlePro-Iso is an integrated system that combines the Esco Versati™ Centrifuge, Esco CO2 incubator, and CelCradle™ benchtop bioreactor system inside the Esco HPI G3. The CelCradle™ system provides cells with an environment of low shear stress, zero foaming, and of high rates of aeration and nutrition, while the CO2 incubator controls the cells surrounding conditions.
The CradlePro-Iso was designed to fully enclose the cell processing system inside an isolator to ensure the safety of the operator, without compromising the quality of the product.
RABS and isolators can be used in the manufacture of biologics, including vaccines, gene therapies, and protein-based drugs. Often, biologic products are preservative-free, contain growth media, and are easily susceptible to contamination. Another area that demands the use of RABS and isolators is the manufacture of sterile drug products with toxic, cytotoxic, and highly potent molecules, which require stringent barriers to protect personnel who are handling these materials. In general, RABS and isolators are being used for smaller-volume and high-value pharmaceuticals. The benefit/cost balance has to be considered when discussing the use of barriers: RABS and isolators come with a high price tag and are associated with additional expenses related to the operation of a cleanroom, such as energy costs, operating costs, testing costs, and gown costs.
Additional cGMP requirements are also included for the commercial production and delivery of these products. These requirements are but not limited to: cleanliness, validation and commissioning, process-material personnel and air flows, cold storage, dry storage, emergency procedures, safety, security, pest control, environmental monitoring, and preventive maintenance.
Reference:
Abdominal Key. (n.d.). Aseptic Manufacturing of Regenerative Medicine Products Using Isolator Technology. Retrieved from: https://abdominalkey.com/aseptic-manufacturing-of-regenerative-medicine-products-using-isolator-technology/
Food and Drug Administration. (2015). Consideration for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products: Guidance for Industry. Retrieved from: https://www.fda.gov/media/106369/download
Harvard Apparatus. (n.d.). Guide to Cell Modification & Re-Engineering Solutions. Retrieved from: https://www.harvardapparatus.com/media/harvard/pdf/Cell%20Modification%20and%20Engineering%20Guide.pdf
Sign up to our newsletter and receive the latest news and updates about our products!






